# Packages
# README
GenFlow: A Bioinformatic WorkFlow for PhyloGenomic Analysis
GenFlow is a streamlined bioinformatic pipeline that combines several tools, including NCBI Datasets command-line, EDirect, Anvi’o, Prodigal, MAFFT, and FastTree, to provide users with an easy, one-command solution for performing phylogenomic and ANI analysis.
Repository
All the necessary data to perform the analysis is available in the GenFlow GitHub repository.
Prerequisites
Make sure you have the following installed:
- Conda for environment management. You can follow this tutorial to install Conda on WSL.
Installation
- Clone the repository:
git clone https://github.com/braddmg/GenFlow.git
cd GenFlow
- Create the Conda environment:
Run the following command to create the environment from the GenFlow.yml file provided:
conda env create -f GenFlow.yml
- Set up the Conda environment:
After creating the environment, run the setup.sh file to install all necessary dependencies and set up the pipeline:
conda activate GenFlow
bash setup.sh
- Reload the Conda environment:
After running the setup.sh script, you will need to reset your Conda environment by deactivating and reactivating it:
conda deactivate
conda activate GenFlow
- Run the GenFlow command to verify installation:
The GenFlow command is now pre-installed as part of the environment. You can check that everything is installed correctly by running the help option:
GenFlow -h
This will show the next message:
Don't worry; sometimes we also don't know what to do.
Syntax: GenFlow [-f|-h|-g|-t|-G|-F|-N|-I]
Options:
-g A txt file containing all accession numbers of reference (g)enomes (default: genomes.txt)
-h Print this (h)elp.
-f Your genomes in (f)asta file format. If you have more than one, provide file names separated by coma:
file1.fasta,file2.fasta,etc. If you do not specify it, GenFlow will use all fasta in the folder.
-t Number of (t)hreads, don't be too rude with your computer.
-G (G)eometric Index value for selecting core genes (default: 0.8).
-F (F)unctional Index value for selecting core genes (default: 0.8).
-N Use this option to work with (N)ucleotide sequences instead of amino acid sequences. It requires a lot of RAM!!
-I Set the (I)nflation value for MCL (default: 2). See https://academic.oup.com/nar/article/30/7/1575/2376029 for more information.
Genome Data
Inside the repository, you'll find a Data folder containing three FASTA files from a potential new subspecies of Aeromonas hydrophila, as well as two sets of reference genomes:
Short Dataset (short.txt): Contains 10 reference genomes of Aeromonas species. We recommend starting with this smaller dataset for your first run.
Large Dataset (large.txt): Contains a larger number of reference genomes for more comprehensive analysis.
You can use either dataset based on your computational capacity and time constraints.
The genome list contain NCBI accession numbers for reference genomes and should look like this:
cat Data/short.txt
GCF_000633175.1
GCF_000820325.1
GCF_016805405.1
GCA_963892115.1
GCF_002850695.3
GCF_022631195.1
GCF_000014805.1
GCF_029025785.1
GCF_002285935.1
GCF_023920205.1
GCF_000820005.1
GCF_028355655.1
Example Command
We suggest using the short dataset to test the pipeline. Running the pipeline on a laptop (in WSL) with 16 GB of RAM and 16 threads takes approximately 1 hour.
Here is an example command to execute the analysis:
cd Data
GenFlow -g short.txt -f INISA09F.fasta,INISA10F.fasta,INISA16F.fasta -t 8 -G 0.8 -F 0.8
In this example:
-g specifies the file containing the accession numbers of the reference genomes.
-f specifies the FASTA files for the analysis.
-t specifies the number of threads to use (recommended 8).
-The -G and -F options specify the geometric and functional index values for core gene selection. In this case, we selected 0.8, which is also the default value for both options.
-As we are not employing the -N flag, the phylogenomic tree will be created with aminoacid sequences. But hey, if you fancy yourself a hardcore evolutionary biologist who swears by DNA sequences, just add the flag :)
-The MCL inflation parameter (-I) was not used, so the default value of 10 was employed. This value is recommended for identifying gene clusters in closely related genomes. Refer to this tutorial for more information: Anvio-pangenomics
Results
Once the analysis completes, you will find the results in a newly created results/ directory.
This folder contains:
Phylogenomic Tree:

The output of the phylogenomic analysis in newick format was visualized in iTOL.
You can observe that the genomes labeled as "INISA" form a distinct clade from the other subspecies. Bootstrap values, based on 1,000 resamplings performed by FastTree, provide support for these clades.
ANI Results:
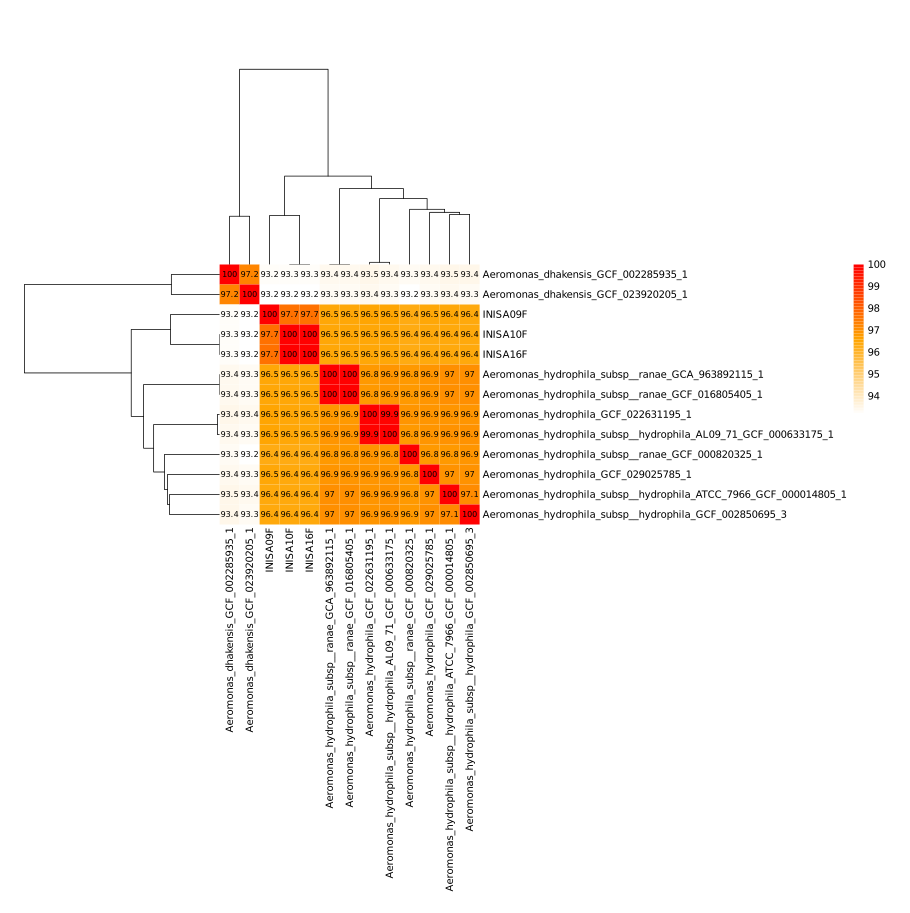
The heatmap, illustrating the results of the Average Nucleotide Identity (ANI) analysis, was created in R using the matrix generated by pyANI. Refer to pyANI for more information.
You will have three different files with different color palettes in the folder.
An important feature implemented in this pipeline is that the FASTA files for the downloaded genomes include both the taxonomy and the accession number in their filenames! This is extremely useful for downstream analysis, but please note that some manual editing may still be required for certain names, as the process isn't entirely perfect (though we did our best :c).
The analysis will conclude with a message notifying you that your phylogenomic plots and ANI results are ready, along with a report of the total time taken for the process.
Logs Folder
The logs folder contains log files generated during the execution of the GenFlow pipeline.
These files are crucial for troubleshooting and understanding the workflow's performance.
To view details about the number of core genes used in the analysis, please check the core.log file.
cat logs/core.log
INFO
===============================================
Your filters resulted in 3222 gene clusters that contain a total of 41886 genes.
for downstream analyses. Just so you know.
In this example, we selected a total of 3,222 genes based on predefined parameters. This large number is due to the analysis being conducted on only 13 genomes, 11 from the same species and two from a closely related species. Thi results in most genes being highly conserved.
Other log files document various steps of the process and can provide insights into any issues encountered during execution.
Citation
If you use GenFlow in your research, please consider citing the following tools:
-
Anvi’o: Eren, A. M., et al. (2015). Anvi'o: An advanced analysis and visualization platform for 'omics data. PeerJ, 3, e1319. https://doi.org/10.7717/peerj.1319
-
MAFFT: Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. https://doi.org/10.1093/molbev/mst010
-
FastTree: Price, M. N., Dehal, P. S., & Arkin, A. P. (2010). FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE, 5(3), e9490. https://doi.org/10.1371/journal.pone.0009490
-
NCBI EDirect: Kans, J. (2013). Entrez Direct: E-utilities on the UNIX command line. In Entrez Programming Utilities Help (Internet). National Center for Biotechnology Information (US). https://www.ncbi.nlm.nih.gov/books/NBK179288/
-
pyANI: Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., & Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Analytical Methods, 8, 12-24. https://doi.org/10.1039/C5AY02550H
-
PRODIGAL: Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., & Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. https://doi.org/10.1186/1471-2105-11-119
Acknowledgments
We acknowledge the use of OpenAI's ChatGPT for assistance in troubleshooting parts of the code.